


Gene Expression Response to Ascorbic Acid in Mice Implanted With Sarcoma S180 Cells
Gene expression response to ascorbic acid in mice
implanted with sarcoma S180 cells
Nina Mikirova and Ruth C. Scimeca
Riordan Clinic, 3100 North Hillside, Wichita, KS, USA
Abstract
In recent years, increasing numbers of studies demonstrated that high-dose ascorbate, which can be achieved by intravenous infusion, has cytotoxic effects on
cancer cells in vitro and in vivo. There are many hypotheses of anti-cancer mechanisms of high dose ascorbate including a pro-oxidative mechanism, inhibition of
angiogenesis and inflammation, enhancement of the anticancer effect of chemotherapy and reduction of chemotherapy-induced side effects. In addition, in recent years there were studies showing that ascorbic acid has effect on gene expression and epigenetic phenomena. In our study we analyzed, by using animal model, the effect of pharmacological concentrations of ascorbic acid on several gene expressions involved in tumorigenesis. To test the effects of ascorbic acid on gene expression, we treated mice with two different concentrations of ascorbic acid after intraperitoneal administration with sarcoma S-180 cells. The injected doses of ascorbic acid were equivalent to 15 g per 70 kg human and 50 g per 70 kg human. Tissue from tumors, liver and kidney was obtained from mice at the end of three weeks of treatment. The gene expression analysis was computed by real time PCR. The results showed significant difference in expression of gene p53 (p<0.02) in tumor tissue between treated and non-treated groups, and reduction of p53 gene expression by size and spreading of tumors. Ascorbate therapy significantly increased expression of NRF2. The experimental data showed that the maximum ascorbate dosage reduced expression of the tumor promoting gene HIF. The dependence of gene expression on the size of tumors was found for P53, HIF and NF-kB. In summary, the results of our study demonstrated that ascorbate therapy had a significant effect on the expression of several genes relevant to the development or inhibition of cancer. Reduced expression of tumor promoting genes as HIF and increased expression of tumor suppression genes such as p53 support the hypothesis that ascorbic acid can act as a potential agent for the suppression of tumor development.
Background
Ascorbic acid (AA, ascorbate, vitamin C) is an essential water
soluble antioxidant that has been studied for decades for its potential
role in preventing chronic diseases [1]. Ascorbate plays a role in limiting
inflammation, regulating cytokine production, and boosting the
immune system [2-8]. It has a variety of properties that have generated
interest in using it against cancer [9-13]: it enhances natural killer cell
activity [14,15], increases collagen synthesis [16], inhibits capillary
tubule formation (angiogenesis) [17,18], reduces inflammation in
cancer patients [3], at millimolar concentrations, shows cytotoxicity
against cancer cells [19-24] and the ability to reduce tumor growth
in vivo [25-34]. Clinical trials to date [35-40] indicate that high dose
(on the order of ten to 100 grams) intravenous ascorbate therapy can
enhance anti-cancer effects of chemotherapy and improve quality of
life in cancer patients.
Recent studies indicate that vitamin C may have important effects
on gene expression. For example, hypoxia inducible factor 1 (HIF-1)
may induce the expression of more than sixty genes, including those
coding for vascular endothelial growth factor (VEGF), erythropoietin
(Epo), and nitric oxide synthase-2 (NOS-2); transcription and
translation of these genes yields proteins that play key roles in
angiogenesis, regulation of glucose metabolism, cell survival, and
cell proliferation [41-44]. Decreased HIF-1 activation is associated
with increased disease free survival in patients with cancer. The
HIF-1a subunit regulates expression of Bcl-2 family proteins, which
in turn protect cells from reactive oxygen species (ROS) induced
apoptosis [45-50]. Since the induction of HIFs may be mediated by
ROS, antioxidants such as vitamin C may inhibit HIF-1 and HIF-1a
expression; this is supported by experimental studies [45-51]. Vitamin
C may also act on HIF-1 expression by inhibiting the expression of
Nuclear Factor kB (NF-kB) genes. NF-kB is involved in inflammation
and tumor development [52-55]. Ascorbate has been shown to inhibit
the expression of NF-kB [52,55-58].
The anticancer effect of ascorbate may be in part due to its ability
to suppress specificity protein (SP) transcription factors (such as
Sp1, Sp3, and Sp4) [59]. Sp-regulated genes are involved in cancer
proliferation (via hepatocyte growth factor receptor, epidermal growth
factor receptor, and cyclin D1), survival (via survivin and Bcl-2) and
angiogenesis (via VEGF and its receptors) [59-62].
Ascorbate may also suppress tumor growth through modulation
of p53 [63-65]. The gene for p53 is particularly important in regulating
cell proliferation, cell cycle progression, DNA repair, senescence and
apoptosis in tumor cells [66-70].
Other genes of importance to cancer that are possibly affected
by millimolar levels of ascorbate include: DNA methyltransferase
(DNMT1), which is responsible for maintaining DNA methylation
and in silencing tumor susceptibility genes [71-76]; human telomerase reverse transcriptase (TERT), which regulates telomere length, can
promote tumor development, increases the anti-apoptotic capacity of
cells, enhances DNA repair [77] and nuclear factor erythroid derived
2 (Nrf2), which has anti-apoptotic effects and may therefore protect
cancer cells [78-79]. Ascorbate has been shown to target the expression
of these genes (promoting p53 while inhibition expression of the
others) in tumor cells [57-59,80], which in turn shifts cells toward the
sub-G1 fraction.
In addition, gene expression studies in vivo and in vitro
have suggested that the carcinostatic effect induced by high
dose concentration ascorbic acid occurred through inhibition of
angiogenesis. In the study [25] the expression of three angiogenesisrelated genes were inhibited by 0.3 times in Fibroblast Growth Factor
(FGF2), 7 times in VEGF and 4 times in Matrix Metallopeptidase 2
(MMP2) of the groups with higher survival rates.
The purpose of the present study is to examine how high dose
AA therapy affects the expression of the several genes in vivo, using
the murine S-180 tumor model, in order to gain more insight into
ascorbates potential mechanisms of action against tumors.
Methods
S180 Cell line
The S180 mouse sarcoma cell line was obtained from ATCC
(Manassas, VA) and cultured in 75 cm2
flasks with 20 mL of RPMI1640 media supplemented with 10% fetal bovine serum (ATCC) and
100 U/ml Penicillin-Streptomycin (Sigma Aldrich, St Louis MO) at
37°C in a 5% CO2
atmosphere.
CD-1 Mice
CD-1 female mice from Charles River were kept under standard
conditions of temperature (22°C) and light (12L/12D) and had access to
water and food (Laboratory Rodent Diet 5001, LabDiet St. Louis MO).
Principles of laboratory animal care following IACUC procedures were
applied, and all experimental protocols were approved by the Ethics
Committee of WSU (Wichita, KS). Mice were given seven days to
acclimate upon arrival, and were weighted and identified on the eight
day. Weight was tracked weekly thereafter and general condition was
recorded three times a week.
Tumor Inoculation and ascorbate therapy
For tumor growth and gene expression experiments, mice were
injected IP with S180 cells. S180 cells were collected by Trypsin
detachment and washed two times by PBS. After detachment 1.5×106
cells diluted in 100 µl PBS were implanted subcutaneously into the right
flank of mice using 25 G needles. One week after tumor inoculation,
ascorbate therapy was commenced via injection. Two AA doses were
tested: “Low AA” was 0.214 mg AA per gram mouse mass (equivalent
to a 15 g dose in a 70 kg human, a typical starting dose in intravenous
ascorbate therapy), and “High AA” 0.714 mg AA per gram mouse mass
(equivalent to a 50 gram dose in a 70 mg human, a maximum dose
typically used in intravenous ascorbate therapy). Ascorbate injections
were administered three times per week. Animals were divided in five
groups of eight mice each:
Group A) Tumor-free mice given the Low AA dose as described
above.
Group B) Tumor-free mice that did not receive ascorbate therapy
(Negative Control)
Group C) S180 inoculated mice given the Low AA dose as described
above.
Group D) S180 inoculated mice given the High AA dose as
described above.
Group E) S180 inoculated mice that did not receive ascorbate
therapy (Positive Control)
Tumor bearing mice were euthanized after three weeks of ascorbate
therapy.
Necropsy and gene expression analysis
Organs (tumors, livers and kidneys) were resected immediately
post-euthanasia. Tumors were measured and weighted at euthanasia.
Samples of kidney, liver and tumor were collected and kept at -80ºC for
RNA extraction. RNA extraction and qRT-PCR analysis were carried
out as follows. Tissues were minced and then passed through 26 gauge
needles to disaggregate. RNA was extracted using TriReagent (SigmaAldrich, Hercules CA) according to manufacturer’s instructions.
Total RNA quality and quantity were evaluated using Nanodrop ND2000 (Thermo Scientific, Pittsburg PA), and 0.5 µg total RNA were
converted to cDNA using iScript RT super-mix in the CFX96 RealTime PCR Detection System (Bio-Rad, Hercules, CA, USA). PCR
reaction conditions consisted of an initial 30 seconds denaturing at
98°C, followed by forty cycles of denaturing at 95°C for 10 seconds,
annealing at 56°C for 15 seconds and extension at 60°C for 15 seconds,
followed by melt-curve analysis. cDNA was quantified using the
Nanodrop ND-2000.
A total of 250 ng cDNA was used to analyze gene-specific
oligonucleotide primers (Table 1) with the SsoAdv Universal SYBR
GREEN Kit. A dissociation curve was run at the end of the reaction
to ensure that only one amplicon was formed and that the amplicons
denatured consistently in the same temperature range for every sample.
cDNA levels were normalized against the reference housekeeping genes
RPS13 and TBP (Table 1) using the genNorm method. The relative
expression of a target gene was computed, based on its real-time PCR
efficiencies (E=2) and the cycle threshold (Ct) difference (Δ) of mean
control versus each sample (ΔCt control−treatment) using RPS13 and
TBP as the reference housekeeping genes.
Statistical analysis
The comparisons of gene expression between different experimental
groups of mice and size of tumor were carried out using the statistic
software Kaleidagraph and Systat software (Inc. Chicago, USA). Data
are presented as medians with IQR and mean ± SE. Differences in
mean values were considered significant at the level of 95% (p<0.05)
(Mann–Whitney U test). The 2−ΔΔCt method was used to calculate the
differences of the expression level of genes [81].
Results and discussion
Tumor growth and necropsy findings
S180 inoculated animals formed abdominal tumors in all
cases except for four mice in Group C (Low AA dose). These four
animals were excluded from subsequent analysis. In all other cases,
S180 inoculations lead to rapid tumor development and noticeable
decrease in animal well-being. One of the mice in Group D (High AA)
developed an encapsulated tumor on the left kidney, coinciding with
an enlargement of the right kidney. Two mice in Group C and two in
Group D developed metastasis in thorax. We did not find any other
significant pathological change in liver or kidney of tumor-bearing
mice.
The weight of mice during the experiment was measured each
week. Tumor-free mice (Group B, “Negative control”) grew at a
relatively steady rate of roughly 0.2 grams/day. In the tumor-bearing
animals (Groups C, D, and E), mouse weight dipped noticeably (14%-
20%) during the first week of therapy, and then rose rapidly to roughly
match that of the tumor-free mice. Intraperitoneal cancer progression
in each group was measured 21 days after tumor cells injections. The
graphical presentation of the weight of the tumors for the each group
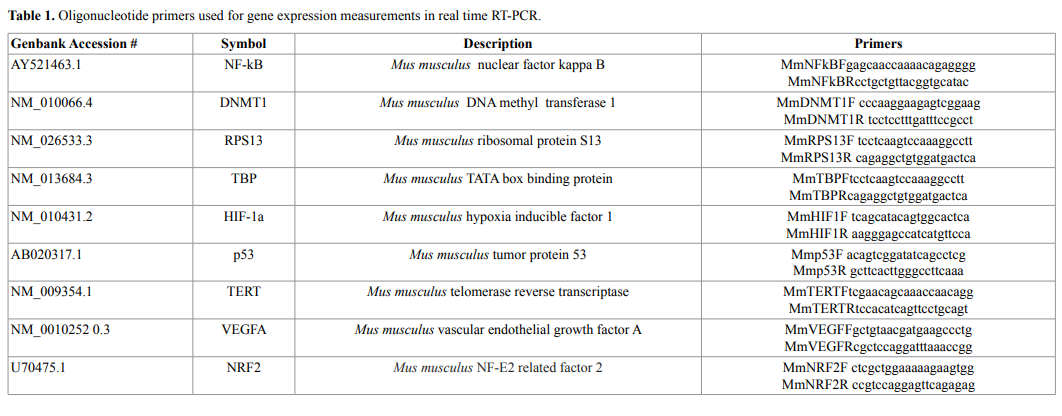
and representative images of the tumors are presented in Figures 1a
and 1b. Only animals that developed tumor are included in Figure 1a.
For untreated mice, the mean tumor volume was 530 ± 162 (SE)
mm3
while the mean volumes for treated mice were 396 ± 105 (SE) mm3
for the Low AA dose and 502 ± 138 (SE) mm3
for the High AA dose.
There was no statistically significant difference between these averages,
suggesting that the ascorbate treatments did not affect tumor size in
this model. The reason for this may be that the injected number of
S180 cells was high that resulted in fast progression of disease, growth
of tumor and deterioration of animals’ conditions. Due to animals’
conditions experiment was stopped 3 weeks after tumor cell injections.
Gene expression on tumor tissue
RNA was isolated from tumors, livers and kidneys of each
mouse and analyzed as described above. Gene expressions in tumors
normalized on the housekeeping genes are presented in Figure 2 and
Table 2 and, while a box plot of gene expression levels for these genes
for two groups of mice (all treated and positive control-non treated) is
shown in Figure 3.
The HIF gene is important in expressing proteins necessary for
tumor angiogenesis, was down-regulated in tumors of mice given
the High AA dose of ascorbate therapy compared to positive control
(Figure 2). This was not statistically significant (p=0.28) but suggests
that further study is warranted. Measured levels of the VEGF gene,
the gene for a key angiogenesis promoter, were too low to draw any
conclusions from. This was also true for the NF-kB gene, where low
levels were obtained and no significant difference between groups was
observed.
Expression of the p53 gene, a key regulator of apoptosis that is
mutated in many tumor cell types, was enhanced roughly two-fold
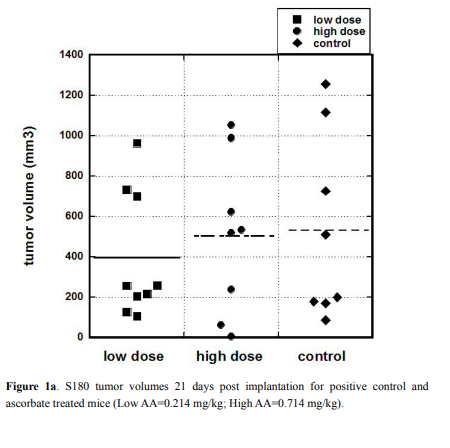
in ascorbate treated tumor cells compared to untreated controls. In
each case (Low AA and High AA), the difference in the mean values
compared to positive controls were statistically significant (p=0.03 and
p=0.04, respectively).
There was also a roughly two-fold increase in NRF2 gene levels in
treated tumor, and this was also statistically significant: comparison
between positive control and Low AA means yielded a p=0.07, while
comparison between control and High AA yielded a p=0.03. When the
treated values were pooled and compared to controls, the mean was
2.5 times higher with a p-value of 0.02. NRF2 up-regulates apoptosis,
so this result, taken with the results for p53, suggests that ascorbate
therapy might increase the likelihood of programed cell death in tumor
cells.
TERT, which affects telomerase, was also elevated in ascorbate
treated tumors (p<0.03 between High AA and control) while DNMT1,

which is involved in DNA methylation, was reduced significantly
(p<0.03) in High AA treated tumors compared to controls. When
results for Low AA and High AA are pooled, we find significant
differences in expression, compared to positive controls, for p53
(p<0.02), NRF2 (p<0.01), TERT (p<0.03), and DNMT1 (p<0.05) using
the Mann–Whitney U test.
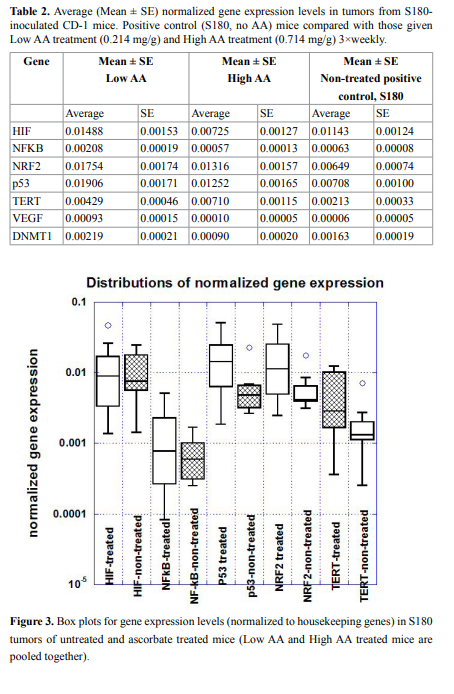
We examined a possible correlation between gene expression and
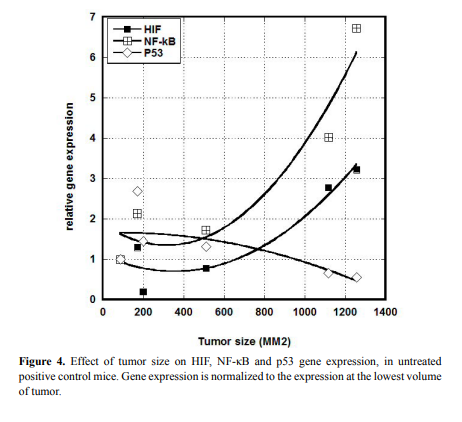
tumor size. Results for HIF, NF-κB, and p53, are shown in Figure 4.
Clearly larger tumors showed elevated levels of HIF and NF-κB
and lower levels of p53 compared to smaller tumors. Using a cut-of of 600 mm3
, we measured the fractional change in expression (from
small to large) for six genes (VEGF was under-expressed in all analyzed
samples and was not included). This is shown in Table 3. For untreated
mice, increases in tumor size increased HIF and NF-κB expression by
roughly 200%, while ascorbate treatments eliminated this effect.
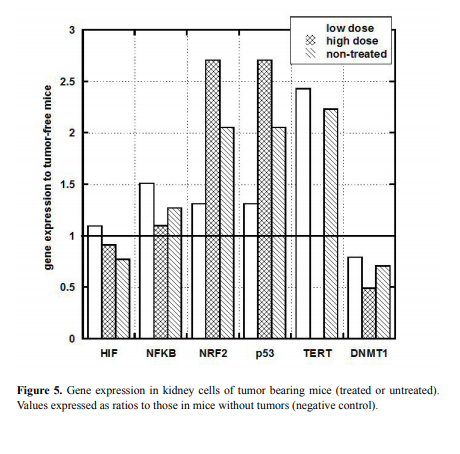
Gene expression in kidney and liver
To investigate the potential effect of tumor development on kidney
and liver gene expression, we analyzed liver and kidney tissue from
tumor bearing and tumor-free mice for gene expression levels. In
particular, the ratio of gene expression in tumor bearing mice to that in
tumor-free mice (Group B-Negative control, as described in Methods)
is shown for kidney in Figure 5.
In liver, gene expression was generally similar to that in tumor-free
mice (data are not shown) and was largely unaffected by treatment. The
exception to this was TERT, which was elevated nearly four-fold (above
tumor-free controls) in untreated mice, but was only mildly elevated in
ascorbate treated mice.
NRF2 and p53 genes measured in kidney tissue were also above
normal in tumor bearing mice, while no noticeable difference was
observed in HIF and NF-kB and a moderate decrease was observed in
DNMT genes.
For untreated tumors, we also examined the effect of tumor size
on kidney gene expression (not shown). For NRF2 and p53 genes, the
relative expression (compared to tumor-free controls) was roughly
3.0 for mice with 200 mm3
tumors, but reduced to roughly 0.9 for
mice with tumors above 600 mm3
. For HIF, relative gene expression
increased with increasing tumor size, rising from roughly 0.5 for mice
with 200 mm3
tumors to values of roughly 1.2 for tumors above mice
with 600 mm3
.
Finally, we examined the effect of metastasis on gene expression.
Several mice developed metastases during this study; for example, three
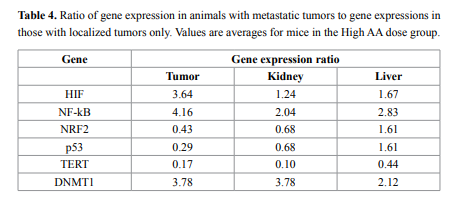
mice in a group of eight given High AA doses developed metastases, as
did two of twelve mice given Low AA treatments. For mice given the
high AA dose, ratios of gene expression (mean) in mice with metastases
to gene expression (mean) for mice without metastases are given in
Table 4. According to these data, in tumor samples of animals that
developed metastasis the HIF, DNMT1, and NF-κB were roughly four
times higher, while p53 and NRF2 genes were 0.3- 0.4 times lower than
in animals with primary tumors only. The same tendency was found in
liver and kidney samples.
Conclusions
We examined the effects of ascorbate therapy in CD1 mice with
aggressive S180 tumors. Our data did not demonstrate a significant
effect of ascorbate on tumor size, but did indicate some effects on gene
expression. Average size of tumors for animals that developed tumor
was 396 ± 300 for group C (low ascorbate), 502 ± 390 for group D (high
ascorbate) and 530 ± 450 for group E (non-treated positive control).
The group treated by high-doses of AA did not show statistical
significant reduction of tumors in comparison with non-treated group
and the average volumes of tumors were similar.
Ascorbate therapy significantly increased expression of tumor
suppressor genes p53.
In group of mice treated by AA the expression of p53 was increased
2.7 fold for low AA treatment and 1.8 fold for group treated by high
dose AA in comparison with non-treated positive control group. The
results of the study have demonstrated that treatment by ascorbic acid
causes to expand p53 tumor suppressive functions.
As ascorbic acid is anti-oxidant and has ability to suppress DNA
damage and genomic instability mediated by reactive oxygen species
(ROS) [91], we expected that HIF gene expression would be affected
by ascorbic acid treatment. The above data show potential effects of
ascorbate on HIF expression. The experimental data showed, that the
maximum ascorbate dose tested (0.714 mg/g) reduced expression
of the tumor promoting gene HIF. The level of mRNA expression
in tumor samples in group treated by this dose of AA was 0.6 fold
lower in comparison with non-treated positive control. There was no
suppression of HIF-1 in group treated by low dose of AA.
We observed that HIF expression is highly dependent upon tumor
size, being over twice as high in mice with large tumors than with mice
bearing smaller tumors. This result correlates with the fact that HIF1 expression is induced by hypoxia and is typically overexpressed in
tumors [82-84]. Expression of HIF was 200% higher in larger tumors
(>600 mm3
) in comparison with smaller tumors (<600 mm3
) for nontreated animals, and the level of mRNA of HIF-1 was decreased for
larger tumors in ascorbic acid treated animals on 60%-80%. As the
result, in mice treated with ascorbate, the increase in HIF expression
with tumor size was suppressed (Table 4).
We also founded that HIF mRNA detected in the liver was reduced
in groups of mice treated with ascorbate, relative to untreated controls
(0.7-0.8 fold).
Our previous research suggests that ascorbate therapy, at the high
doses associated with intravenous infusions clinically, can inhibit
angiogenesis and reduce tumor inflammation [3,4,17,18]. In study
[92], lung blood vessel proliferation and the incidence of pulmonary
malignant tumors were reduced in the offspring of mice expose to
cigarette smoke and a preventive treatment with ascorbic acid in
drinking water throughout the pregnancy. We expected therefore,
to see the effects on HIF genes involved in angiogenesis promotion,
and the gene for NF-κB, a critical factor in inflammation. We did not
observe systematic effect of ascorbate therapy on NF-κB. One possible
explanation is that our tumor model, with over 106
S180 cells injected
into the mice, was too fast acting and too aggressive for ascorbate
therapy to slow down or significantly alter. The other possibility was
that ascorbic acid levels in tumors did not reach pharmacological
concentrations capable to produce the expected results in such a
short time acting only as an antioxidant ROS scavenger as reported
in study [93]. Tumors varied substantially in size, but grew so quickly
and had such a deleterious effect on animal health that our animals
needed to be euthanized after only a few weeks. Our hope for future
experiment would be to use a slower growing tumor model, in which
ascorbate therapy can be tested over a longer period of time. Another
approximation would be to analyze the combination of ascorbic acid
with a synergistic compound as in study [94] to evaluate the effect on
tumor development and gene expression.
However, the dependence of gene expression on the size of tumors
and the effect of ascorbic acid treatment on gene expression were
found for NK-κB (Table 3). NF-κB expression was increased for larger
tumors in positive control group on 200% and decreased in ascorbic
acid treated animals on average 25%.
In tumor bearing mice, p53 gene expression was elevated in
kidneys, but normal in liver. Compared to untreated tumor-bearing
mice, we found a statistically significant increase in expression of
gene p53 (p<0.02) in tumor-bearing mice treated with ascorbate. This
is consistent with reports in the literature of ascorbate’s effect on the
p53 gene [30,64,80]. Our data showed that p53 gene expression was
elevated in ascorbate treated groups of mice with tumors, tended to
decrease as tumors became larger and was reduced in animals with
metastatic tumors. Taken together, the data suggest that p53 gene
expression decreases in large and metastatic tumors, but can be upregulated by ascorbate therapy.
The role of p53 as a tumor suppressor has been extensively studied
[66-70]. It is estimated that roughly half of all tumor cell types harbor
mutations in the p53 gene, and in most of the remaining cancers the
gene is inactivated by a variety of mechanisms. Reactivation of p53 is
an important strategy for inhibiting tumor growth and proliferation. It
is typically activated in response to stress signals and transcriptionally
induces a lot of target genes relevant to cell cycle progression, DNA
repair, apoptosis, and tumor cell metabolism. The evidence presented
above, that ascorbate may activate p53 expression in tumor cells, is
encouraging and warrants further study.
Ascorbic acid treatment increased expression of NRF2 in both
groups of treated animals in comparison with non-treated group. The
role of NRF2 in carcinogenesis and cancer is still actively disputed and
remains unresolved. There is abundant evidence that activation of
NRF2 can suppress carcinogenesis, especially in its earliest stages. The
current thinking is that NRF2 activity may be desirable in early stages
of tumorigenesis, when the host is seeking to control premalignant
carcinogenesis, but may be undesirable in later stages, when it could
make fully malignant cancer cells become resistant to treatment
[85,86]. In our model, with treatments beginning one week after tumor
inoculation, the tumors may be already established, so that the analysis
of the changes in NRF2 expression accompanying ascorbate therapy
might have been more beneficial if the treatments were started earlier.
Comparison of the expression of DNMT1 in treated by AA and
non-treated groups showed that high dose AA downregulated DNMT1
in tumor tissue 0.5 folds and there was no suppression by low dose AA.
The same result was found for kidney tissues.
Expression of DNMTs in cancer and DNA methylation
patterns in tumor cells in comparison to those of normal cells were
analyzed in numerous studies and it was postulated that in most of
the carcinogenesis DNMTs are over expressed [87-90]. Excessive
amounts of DNMT1 may participate in the de novo methylation of
CpG islands that are not methylated in normal cells and contribute to
tumor development through CpG island methylation-mediated gene
inactivation. Interestingly, two converse trends of DNA methylation
changes were observed in many tumors. On the one hand, promoters
of tumor suppressor genes are often hypermethylated leading to the
silencing of the genes. On the other hand, the activity of DNMT1 is
increased leading to DNA hypermethylation. Inactivation of tumor
suppressor genes is central to the development of all common forms
of human cancer.
TERT is involved in telomerase, which is thought to aid tumor
cell growth and allow tumor cells to become immortal, so that TERT
expression is considered a potential target for anticancer therapy.
In our study, expression of TERT was increased in tumor tissue for
treated groups in comparison with non-treated group (2.0 folds for low
dose AA and 3.3 for high dose AA). In kidney tissue overexpression of
TERT was not found and there was suppression of TERT in liver tissue
for groups treated by AA (0.5 folds for low dose and 0.45 fold for high
dose).
Comparison of the gene expression in mice that developed
metastasis with mice having localized tumors, showed that in animals
that developed metastasis the HIF and NF-κB were 3.6-4 times higher,
p53 and NRF2 were 0.4 and 0.3 times lower and DNMT1 was 3.8 times
higher. The same tendency was found in liver and kidney samples of
these two groups of mice.
In summary, the results of our study demonstrated that ascorbate
therapy had effect on the expression of several genes relevant to the
development or inhibition of cancer. Reduced expression of such
tumor promoting genes as HIF and increased expression of tumor
suppression genes such as p53 support the hypothesis that AA can
act as potential agents for the suppression of tumor development.
To obtain a better knowledge of the AA effect in gene expression and
tumor development and metastasis, further studies should be done
with less aggressive course of tumor development and a larger number
of cancer related genes.
Acknowledgements
The study was supported by Flossie E West Memorial Trust.
References
1. Levy TE (2012) Primal panacea. MedFox Publishing
2. Hartel C, Puzik A, Gopel W, Temming P, Bucsky P, et al. (2007) Immunomodulatory
effect of vitamin C on intracytoplasmic cytokine production in neonatal cord blood
cells. Neonatology 91: 54-60. [Crossref]
3. Mikirova N, Casciari J, Rogers A, Taylor P (2012) Effect of high-dose intravenous
vitamin C on inflammation in cancer patients. J Transl Med 10: 189. [Crossref]
4. Mikirova N, Rogers A, Casciari J, Taylor P (2012) Effect of high dose intravenous
ascorbic acid on the level of inflammation in patients with rheumatoid arthritis. Modern
Research in Inflammation 1: 26-32.
5. Ichim TE, Minev B, Braciak T, Luna B, Hunninghake R, et al. (2011) Intravenous
ascorbic acid to prevent and treat cancer-associated sepsis? J Transl Med 9: 25.
[Crossref]
6. Härtel C, Strunk T, Bucsky P, Schultz C (2004) Effects of vitamin C on intracytoplasmic
cytokine production in human whole blood monocytes and lymphocytes. Cytokine 27:
101-106. [Crossref]
7. de la Fuente M, Ferrández MD, Burgos MS, Soler A, Prieto A, et al. (1998) Immune
function in aged women is improved by ingestion of vitamins C and E. Can J Physiol
Pharmacol 76: 373-380. [Crossref]
8. Vojdani A, Ghoneum M (1993) In vivo effect of ascorbic acid on enhancement of
human natural killer cell activity. Nutr Res 13: 753–754.
9. Ohno S, Ohno Y, Suzuki N, Soma G, Inoue M (2009) High-dose vitamin C (ascorbic
acid) therapy in the treatment of patients with advanced cancer. Anticancer Res 29:
809-815. [Crossref]
10. Parrow NL, Leshin JA, Levine M (2013) Parenteral Ascorbate As a Cancer Therapeutic:
A Reassessment Based on Pharmacokinetics. Antioxid Redox Signal 19: 2141-2156.
[Crossref]
11. Du J, Cullen JJ, Buettner GR (2012) Ascorbic acid: chemistry, biology and the
treatment of cancer. Biochim Biophys Acta 1826: 443-457. [Crossref]
12. Jacobs C, Hutton B, Ng T, Shorr R, Clemons M (2015) Is there a role for oral or
intravenous ascorbate (vitamin C) in treating patients with cancer? A systematic
review. Oncologist 20: 210-223. [Crossref]
13. McCORMICK WJ (1954) Cancer: the preconditioning factor in pathogenesis; a new
etiologic approach. Arch Pediatr 71: 313-322. [Crossref]
14. Herberman R (1983) Possible role of natural killer cells in host resistance against
tumors and diseases. Clin Immunol Allergy 3: 479-485.
15. Wilson MK, Baguley BC, Wall C, Jameson MB, Findlay MP (2014) Review of highdose intravenous vitamin C as an anticancer agent. Asia Pac J Clin Oncol 10: 22-37.
[Crossref]
16. Riordan NH, Riordan HD, Meng X, Li Y, Jackson JA (1995) Intravenous ascorbate as
a tumor cytotoxic chemotherapeutic agent. Med Hypotheses 44: 207-213. [Crossref]
17. Mikirova NA, Ichim TE, Riordan NH (2008) Anti-angiogenic effect of high doses of
ascorbic acid. J Transl Med 6: 50. [Crossref]
18. Mikirova NA, Casciari JJ, Riordan NH (2010) Ascorbate inhibition of angiogenesis
in aortic rings ex vivo and subcutaneous Matrigel plugs in vivo. J Angiogenes Res 2:
2. [Crossref]
19. Casciari JJ, Riordan NH, Schmidt TL, Meng XL, Jackson JA, et al. (2001) Cytotoxicity
of ascorbate, lipoic acid, and other antioxidants in hollow fibre in vitro tumours. Br J
Cancer 84: 1544-1550. [Crossref]
20. Chen Q, Espey MG, Krishna MC, Mitchell JB, Corpe CP, et al. (2005) Pharmacologic
ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver
hydrogen peroxide to tissues. Proc Natl Acad Sci U S A 102: 13604-13609. [Crossref]
21. Du J, Martin SM, Levine M, Wagner BA, Buettner GR, et al. (2010) Mechanisms
of ascorbate-induced cytotoxicity in pancreatic cancer. Clin Cancer Res 16: 509-520.
[Crossref]
22. Chen Q, Espey MG, Sun AY, Lee JH, Krishna MC, et al. (2007) Ascorbate in
pharmacologic concentrations selectively generates ascorbate radical and hydrogen
peroxide in extracellular fluid in vivo. Proc Natl Acad Sci USA 104: 8749–8754.
[Crossref]
23. Sinnberg T, Noor S, Venturelli S, Berger A, Schuler P, et al. (2014) The ROS-induced
cytotoxicity of ascorbate is attenuated by hypoxia and HIF-1alpha in the NCI60 cancer
cell lines. J Cell Mol Med 18: 530-541. [Crossref]
24. Chen Q, Espey MG, Sun AY, Pooput C, Kirk KL, et al. (2008) Pharmacologic doses
of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in
mice. Proc Natl Acad Sci U S A 105: 11105-11109. [Crossref]
25. Yeom CH, Lee G, Park JH, Yu J, Park S, et al. (2009) High dose concentration
administration of ascorbic acid inhibits tumor growth in BALB/C mice implanted
with sarcoma 180 cancer cells via the restriction of angiogenesis. J Transl Med 7: 70.
[Crossref]
26. Pollard HB, Levine MA, Eidelman O, Pollard M (2010) Pharmacological ascorbic acid
suppresses syngeneic tumor growth and metastases in hormone-refractory prostate
cancer. In Vivo 24: 249-255. [Crossref]
27. Frömberg A, Gutsch D, Schulze D, Vollbracht C, Weiss G, et al. (2011) exerts anti-proliferative effects through cell cycle inhibition and sensitizes tumor cells
towards cytostatic drugs. Cancer Chemother Pharmacol 67: 1157-1166. [Crossref]
28. Campbell A, Jack T (1979) Acute reactions to mega ascorbic acid therapy in malignant
disease. Scott Med J 24: 151-153. [Crossref]
29. Prasad KN, Hernandez C, Edwards-Prasad J, Nelson J, Borus T, et al. (1994)
Modification of the effect of tamoxifen, cis-platin, DTIC, and interferon-alpha 2b on
human melanoma cells in culture by a mixture of vitamins. Nutr Cancer 22: 233-245.
[Crossref]
30. Reddy VG, Khanna N, Singh N (2001) Vitamin C augments chemotherapeutic
response of cervical carcinoma HeLa cells by stabilizing P53. Biochem Biophys Res
Commun 282: 409-415. [Crossref]
31. Prasad KN, Sinha PK, Ramanujam M, Sakamoto A (1979) Sodium ascorbate
potentiates the growth inhibitory effect of certain agents on neuroblastoma cells in
culture. Proc Natl Acad Sci U S A 76: 829-832. [Crossref]
32. Espey MG, Chen P, Chalmers B, Drisko J, Sun AY, et al. (2011) Pharmacologic
ascorbate synergizes with gemcitabine in preclinical models of pancreatic cancer. Free
Radic Biol Med 50: 1610-1619. [Crossref]
33. Abdel-Latif MM, Raouf AA, Sabra K, Kelleher D, Reynolds JV (2005) Vitamin C
enhances chemosensitization of esophageal cancer cells in vitro. J Chemother 17: 539-
549. [Crossref]
34. Verrax J, Calderon PB (2009) Pharmacologic concentrations of ascorbate are achieved
by parenteral administration and exhibit antitumoral effects. Free Radic Biol Med 47:
32-40. [Crossref]
35. Ma Y, Chapman J, Levine M, Polireddy K, Drisko J, et al. (2014) High-dose parenteral
ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of
chemotherapy. Sci Transl Med 6: 222ra18. [Crossref]
36. Stephenson CM, Levin RD, Spector T, Lis CG (2013) Phase I clinical trial to evaluate
the safety, tolerability, and pharmacokinetics of high-dose intravenous ascorbic acid in
patients with advanced cancer. Cancer Chemother Pharmacol 72: 139–146. [Crossref]
37. Monti DA, Mitchell E, Bazzan AJ, Littman S, Zabrecky G, et al. (2012) Phase I
evaluation of intravenous ascorbic acid in combination with gemcitabine and erlotinib
in patients with metastatic pancreatic cancer. PLoS One 7: e29794. [Crossref]
38. Riordan HD, Casciari JJ, González MJ, Riordan NH, Miranda-Massari JR, et al. (2005)
A pilot clinical study of continuous intravenous ascorbate in terminal cancer patients. P
R Health Sci J 24: 269-276. [Crossref]
39. Welsh JL, Wagner BA, van’t Erve TJ, Zehr PS, Berg DJ, et al. (2013) Pharmacological
ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic
cancer (PACMAN): results from a phase I clinical trial. Cancer Chemother Pharmacol
71: 765-775. [Crossref]
40. Vollbracht C, Schneider B, Leendert V, Weiss G, Auerbach L, et al. (2011)
Intravenous vitamin C administration improves quality of life in breast cancer patients
during chemo-/radiotherapy and aftercare: Results of a retrospective, multicentre,
epidemiological cohort study in Germany. In Vivo 25: 983-990. [Crossref]
41. Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3: 721-732.
[Crossref]
42. Semenza GL (2001) HIF-, O(2), and the 3 PHDs: how animal cells signal hypoxia to
the nucleus. Cell 107: 1-3. [Crossref]
43. Kimura H, Weisz A, Ogura T, Hitomi Y, Kurashima Y, et al. (2001) Identification of
hypoxia-inducible factor 1 ancillary sequence and its function in vascular endothelial
growth factor gene induction by hypoxia and nitric oxide. J Biol Chem 276: 2292-2298.
[Crossref]
44. Brennan PA, Umar T, Smith GI, Lo CH, Tant S (2002) Expression of nitric oxide
synthase-2 in cutaneous squamous cell carcinoma of the head and neck. Br J Oral
Maxillofac Surg 40: 191-194. [Crossref]
45. Wang Y, Liu Y, Malek SN, Zheng P, Liu Y (2011) Targeting HIF1α eliminates cancer
stem cells in hematological malignancies. Cell Stem Cell 8: 399-411. [Crossref]
46. Deeb G, Vaughan MM, McInnis I, Ford LA, Sait SN, et al. (2011) Hypoxia-inducible
factor-1α protein expression is associated with poor survival in normal karyotype adult
acute myeloid leukemia. Leuk Res 35: 579-584. [Crossref]
47. Wellmann S, Guschmann M, Griethe W, Eckert C, von Stackelberg A, et al. (2004)
Activation of the HIF pathway in childhood ALL, prognostic implications of VEGF.
Leukemia 18: 926-933. [Crossref]
48. Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, et al. (2012) Anti-apoptotic Mcl-1 is
essential for the development and sustained growth of acute myeloid leukemia. Genes
Dev 26: 120-125. [Crossref]
49. Del Principe MI, Del Poeta G, Venditti A, Buccisano F, Maurillo L, et al. (2005)
Apoptosis and immaturity in acute myeloid leukemia. Hematology 10: 25-34. [Crossref]
50. Broome HE, Yu AL, Diccianni M, Camitta BM, Monia BP, et al. (2002) Inhibition
of Bcl-xL expression sensitizes T-cell acute lymphoblastic leukemia cells to
chemotherapeutic drugs. Leuk Res 26: 311-316. [Crossref]
51. Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, et al. (2007) HIF-dependent antitumorigenic
effect of antioxidants in vivo. Cancer Cell 12: 230-238. [Crossref]
52. Braun T, Carvalho G, Fabre C, Grosjean J, Fenaux P, et al. (2006) Targeting NFkappaB in hematologic malignancies. Cell Death Differ 13: 748-758. [Crossref]
53. Karin M, Cao Y, Greten FR, Li ZW (2002) NF-kappaB in cancer: from innocent
bystander to major culprit. Nat Rev Cancer 2: 301-310. [Crossref]
54. Gilmore TD, Koedood M, Piffat KA, White DW (1996) Rel/NF-kappaB/IkappaB
proteins and cancer. Oncogene 13: 1367-1378. [Crossref]
55. Aggarwal BB, Sethi G, Nair A, Ichikawa H (2006) Nuclear Factor-kB: A Holy Grail
in Cancer Prevention and Therapy. Current Signal Transduction Therapy 1: 25-52.
56. Reikvam H, Olsnes AM, Gjertsen BT, Ersvar E, Bruserud Ø (2009) Nuclear factorkappaB signaling: a contributor in leukemogenesis and a target for pharmacological
intervention in human acute myelogenous leukemia. Crit Rev Oncog 15: 1-41.
[Crossref]
57. Cárcamo JM, Pedraza A, Bórquez-Ojeda O, Zhang B, Sanchez R, et al. (2004) Vitamin
C is a kinase inhibitor: dehydroascorbic acid inhibits IkappaBalpha kinase beta. Mol
Cell Biol 24: 6645-6652. [Crossref]
58. Bowie AG, O’Neill LA (2000) Vitamin C inhibits NF-kappa B activation by TNF via
the activation of p38 mitogen-activated protein kinase. J Immunol 165: 7180-7188.
[Crossref]
59. Pathi SS, Lei P, Sreevalsan S, Chadalapaka G, Jutooru I, et al. (2011) Pharmacologic
doses of ascorbic acid repress specificity protein (Sp) transcription factors and Spregulated genes in colon cancer cells. Nutr Cancer 63: 1133-1142. [Crossref]
60. Jutooru I, Chadalapaka G, Sreevalsan S, Lei P, Barhoumi R, et al. (2010) Arsenic
trioxide downregulates specificity protein (Sp) transcription factors and inhibits bladder
cancer cell and tumor growth. Exp Cell Res 316: 2174-2188. [Crossref]
61. Jutooru I, Chadalapaka G, Lei P, Safe S (2010) Inhibition of NFkappaB and pancreatic
cancer cell and tumor growth by curcumin is dependent on specificity protein downregulation. J Biol Chem 285: 25332-25344. [Crossref]
62. Jutooru I, Chadalapaka G, Abdelrahim M, Basha MR, Samudio I, et al. (2010) Methyl
2-cyano-3,12-dioxooleana-1,9-dien-28-oate decreases specificity protein transcription
factors and inhibits pancreatic tumor growth: role of microRNA- 27a. Mol Pharmacol
78: 226-236. [Crossref]
63. Hahm E, Jin DH, Kang JS, Kim YI, Hong SW, et al. (2007) The molecular mechanisms
of vitamin C on cell cycle regulation in B16F10 murine melanoma. J Cell Biochem 102:
1002-1010. [Crossref]
64. Kim JE, Jin DH, Lee SD, Hong SW, Shin JS, et al. (2008) Vitamin C inhibits p53-
induced replicative senescence through suppression of ROS production and p38 MAPK
activity. Int J Mol Med 22: 651-655. [Crossref]
65. Lee SK, Kang JS, Jung da J, Hur DY, Kim JE, et al. (2008) Vitamin C suppresses
proliferation of the human melanoma cell SK-MEL-2 through the inhibition of
cyclooxygenase- (COX-2) expression and the modulation of insulin-like growth factor
II (IGF-II) production. J Cell Physiol 216: 180-188. [Crossref]
66. Muller PAJ, Vousden KH (2013) p53 mutations in cancer. Nature Cell Biology 15: 1-8.
67. Riley T, Sontag E, Chen P, Levine A (2008) Transcriptional control of human p53-
regulated genes. Nat Rev Mol Cell Biol 9: 402-412. [Crossref]
68. Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408: 307-
310. [Crossref]
69. Vousden KH, Lane DP (2007) p53 in health and disease. Nat Rev Mol Cell Biol 8:
275-283. [Crossref]
70. Marchenko ND, Moll UM (2007) The role of ubiquitination in the direct mitochondrial
death program of p53. Cell Cycle 6: 1718-1723. [Crossref]
71. Bashtrykov P, Jeltsch A. (2015) DNMT1-associated DNA methylation changes in
cancer. 2015. Cell Cycle 14: 1-5. [Crossref]
72. Robertson KD (2005) DNA methylation and human disease. Nat Rev Genet 6: 597-610.
[Crossref]
73. Costello JF, Frühwald MC, Smiraglia DJ, Rush LJ, Robertson GP, et al. (2000)
Aberrant CpG-island methylation has non-random and tumour-type-specific patterns.
Nat Genet 24: 132-138. [Crossref]
74. Chan AO, Broaddus RR, Houlihan PS, Issa JP, Hamilton SR, et al. (2002) CpG island
methylation in aberrant crypt foci of the colorectum. Am J Pathol 160: 1823-1830.
[Crossref]
75. Laird PW (2003) The power and the promise of DNA methylation markers. Nat Rev
Cancer 3: 253-266. [Crossref]
76. Baylin SB, Makos M, Wu JJ, Yen RW, de Bustros A, et al. (1991) Abnormal patterns of
DNA methylation in human neoplasia: potential consequences for tumor progression.
Cancer Cells 3: 383-390. [Crossref]
77. Qin Y, Guo H, Tang B, Yang SM (2014) The non-reverse transcriptase activity of the
human telomerase reverse transcriptase promotes tumor progression (Review). Int J
Oncol 45: 525-531. [Crossref]
78. Sporn MB, Liby KT (2012) NRF2 and cancer: the good, the bad and the importance of
context. Nat Rev Cancer 12: 564-571. [Crossref]
79. Kensler TW, Wakabayashi N (2010) Nrf2: friend or foe for chemoprevention?
Carcinogenesis 31: 90-99. [Crossref]
80. Venturelli S, Sinnberg TW, Berger A, Noor S, Levesque MP3, et al. (2014) Epigenetic
impacts of ascorbate on human metastatic melanoma cells. Front Oncol 4: 227.
[Crossref]
81. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using realtime quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402-408.
[Crossref]
82. Semenza GL (1999) Regulation of mammalian O2 homeostasis by hypoxia-inducible
factor 1. Annu Rev Cell Dev Biol 15: 551-578. [Crossref]
83. Zhong H, DeMarzo AM, Laughner E, LimM, HiltonDA, et al. (1999) Overexpression
of hypoxia-inducible factor 1alpha in common human cancers and their metastases.
Cancer Res 59: 5830-5835. [Crossref]
84. Talks KL, Turley H, Gatter KC, Maxwell PH, PughCW, et al. (2000) The expression
and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal
human tissues, cancers, and tumor-associated macrophages. Am J Pathol 157: 411-421.
[Crossref]
85. Hayes JD, McMahon M, Chowdhry S, Dinkova-Kostova AT (2010) Cancer
chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid
Redox Signal 13: 1713-1748. [Crossref]
86. Hu R, Saw CL, Yu R, Kong AN (2010) Regulation of NF-E2-related factor 2 signaling
for cancer chemoprevention: antioxidant coupled with antiinflammatory. Antioxid.
Redox Signal 13: 1679-1698.
87. Robertson KD, Uzvolgyi E, Liang G, Talmadge C, Sumegi J, et al. (1999) The human
DNA methyltransferases (DNMTs), 3a and 3b: coordinate mRNA expression in normal
tissues and overexpression in tumors. Nucleic Acids Res 27: 2291-2298. [Crossref]
88. Siegfried Z, Cedar H (1997) DNA methylation: a molecular lock. Curr Biol 7: R305-
307. [Crossref]
89. Robertson KD, Jones PA (2000) DNA methylation: past, present and future directions.
Carcinogenesis 21: 461-467. [Crossref]
90. Tycko B (2000) Epigenetic gene silencing in cancer. J Clin Invest 105: 401-407.
[Crossref]
91. Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, et al. (2007) HIF-dependent antitumorigenic
effect of antioxidants in vivo. Cancer Cell 12: 230-238. [Crossref]
92. De Flora S, Ganchev G, Iltcheva M, La Maestra S, Micale RT, et al. (2016)
Pharmacological Modulation of Lung Carcinogenesis in Smokers: Preclinical and
Clinical Evidence. Trends Pharmacol Sci 37: 120-142. [Crossref]
93. Kumar A, Singh B, Sharma PR, Bharate SB, Saxena AK, et al. (2016) A novel
microtubule depolymerizing colchicine analogue triggers apoptosis and autophagy in
HCT-116 colon cancer cells. Cell Biochem Funct 34: 69-81. [Crossref]
94. Nayak D, Minz AP, Ashe S, Rauta PR, Kumari M, et al. (2016) Synergistic combination
of antioxidants, silver nanoparticles and chitosan in a nanoparticle based formulation:
Characterization and cytotoxic effect on MCF-7 breast cancer cell lines. J Colloid
Interface Sci 470: 142-152. [Crossref]



